![]()


LaminDB - A data framework for biology¶
Makes your data queryable, traceable, reproducible, and FAIR. One API: lakehouse, lineage, feature store, ontologies, LIMS, ELN.
Why?
Reproducing analytical results or understanding how a dataset or model was created can be a pain. Training models on historical data, LIMS & ELN systems, orthogonal assays, or datasets from other teams is even harder. Even maintaining an overview of a project’s datasets & analyses is more difficult than it should be.
Biological datasets are typically managed with versioned storage systems, GUI-focused platforms, structureless data lakes, rigid data warehouses (SQL, monolithic arrays), or tabular lakehouses.
LaminDB extends the lakehouse architecture to biological registries & datasets beyond tables (DataFrame, AnnData, .zarr, .tiledbsoma, …).
It provides enough structure to enable queries across many datasets, enough freedom to keep the pace of R&D high, and rich context in form of data lineage and metadata for humans and AI.
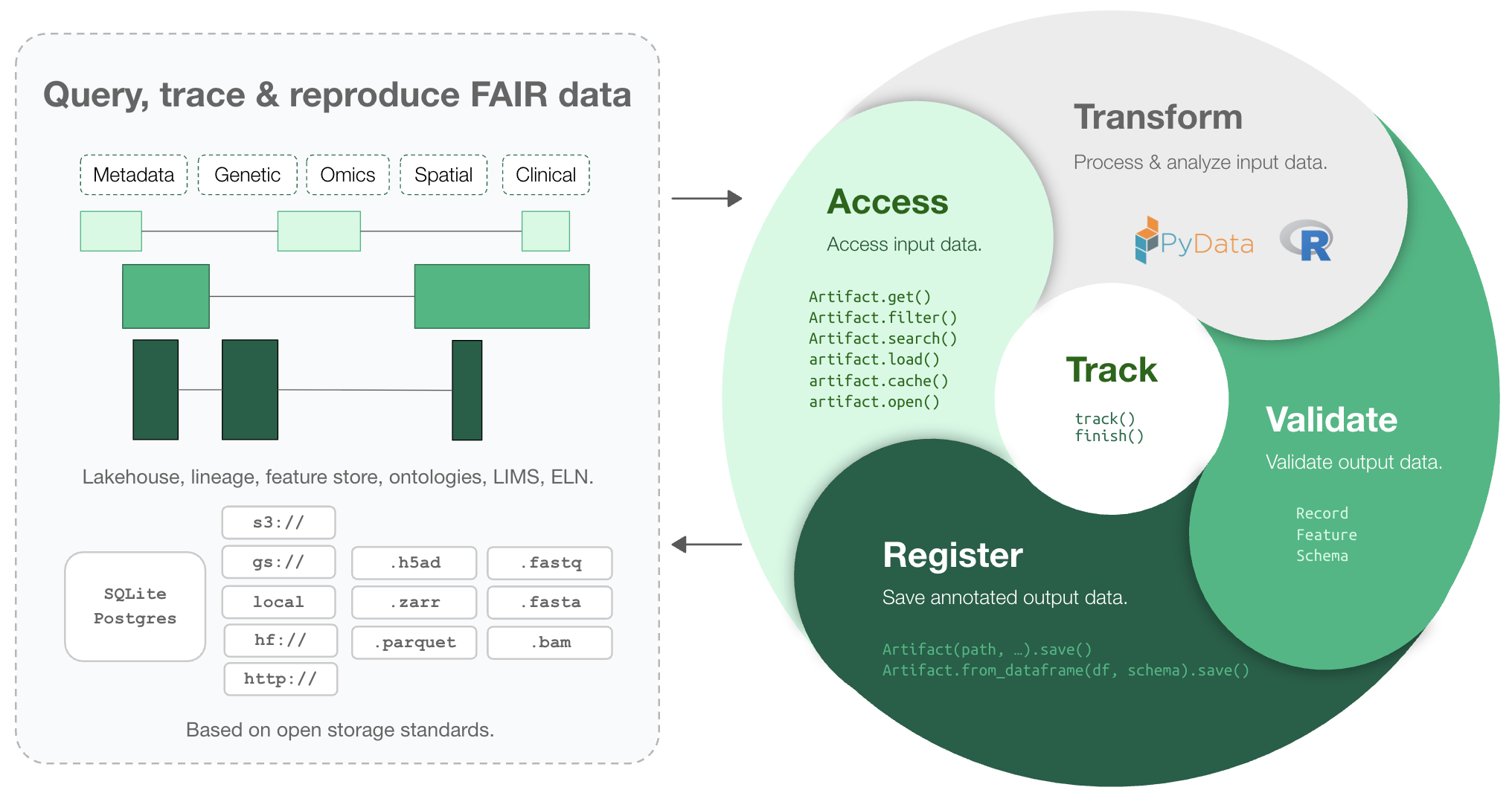
Highlights:
lineage → track inputs & outputs of notebooks, scripts, functions & pipelines with a single line of code
lakehouse → manage, monitor & validate schemas; query across many datasets
feature store → manage features & labels; leverage batch loading
FAIR datasets → validate & annotate
DataFrame,AnnData,SpatialData,parquet,zarr, …LIMS & ELN → manage experimental metadata, ontologies & markdown notes
unified access → storage locations (local, S3, GCP, …), SQL databases (Postgres, SQLite) & ontologies
reproducible → auto-version & timestamp execution reports, source code & environments
zero lock-in & scalable → runs in your infrastructure; not a client for a rate-limited REST API
extendable → create custom plug-ins based on the Django ORM
If you want a GUI: LaminHub is a data collaboration hub built on LaminDB similar to how GitHub is built on git.
Who uses it?
Scientists & engineers in pharma, biotech, and academia, including:
Pfizer – A global BigPharma company with headquarters in the US
Ensocell Therapeutics – A BioTech with offices in Cambridge, UK, and California
DZNE – The National Research Center for Neuro-Degenerative Diseases in Germany
Helmholtz Munich – The National Research Center for Environmental Health in Germany
scverse – An international non-profit for open-source omics data tools
The Global Immunological Swarm Learning Network – Research hospitals at U Bonn, Harvard, MIT, Stanford, ETH Zürich, Charite, Mount Sinai, and others
Docs¶
Copy summary.md into an LLM chat and let AI explain or read the docs.
Setup¶
Install the Python package:
pip install lamindb
Create a LaminDB instance:
lamin init --modules bionty --storage ./quickstart-data # or s3://my-bucket, gs://my-bucket
Or if you have write access to an instance, connect to it:
lamin connect account/name
Quickstart¶
Lineage¶
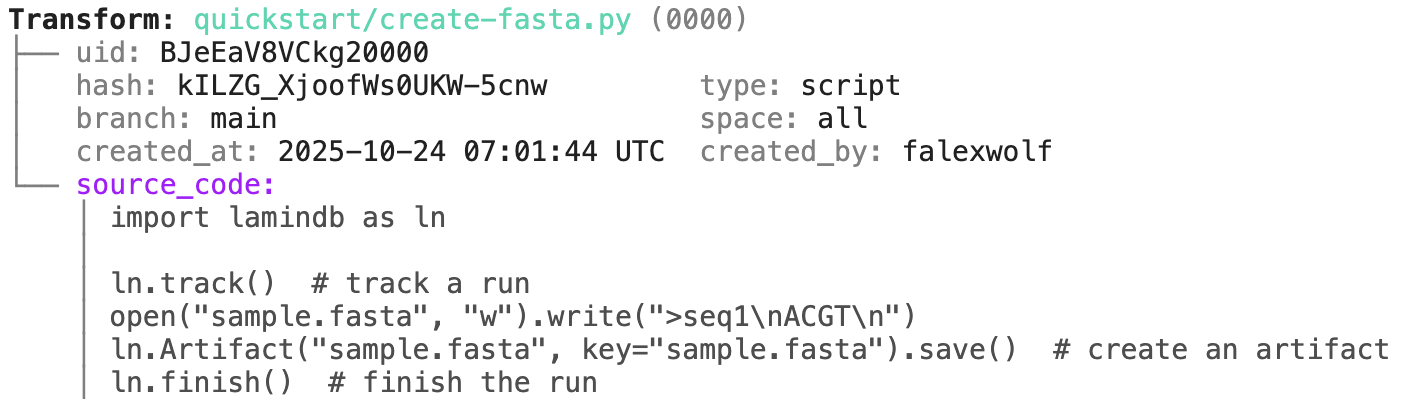
Create a dataset while tracking source code, inputs, outputs, logs, and environment:
import lamindb as ln
ln.track() # track execution of source code as a run
open("sample.fasta", "w").write(">seq1\nACGT\n") # create a dataset
ln.Artifact("sample.fasta", key="sample.fasta").save() # save dataset as an artifact
ln.finish() # mark the run as finished
→ connected lamindb: anonymous/test-transfer
→ created Transform('5UreeBqnzjaw0000', key='README.ipynb'), started new Run('lwMrMpWqk4EjwXmZ') at 2025-11-27 13:42:58 UTC
→ notebook imports: anndata==0.12.2 bionty==1.9.1 lamindb==1.17a1 numpy==2.3.5 pandas==2.3.3
• recommendation: to identify the notebook across renames, pass the uid: ln.track("5UreeBqnzjaw")
! calling anonymously, will miss private instances
! cells [(0, 2), (16, 18)] were not run consecutively
→ finished Run('lwMrMpWqk4EjwXmZ') after 3s at 2025-11-27 13:43:01 UTC
Running this snippet as a script (python create-fasta.py) produces the following data lineage:
artifact = ln.Artifact.get(key="sample.fasta") # get artifact by key
artifact.view_lineage()

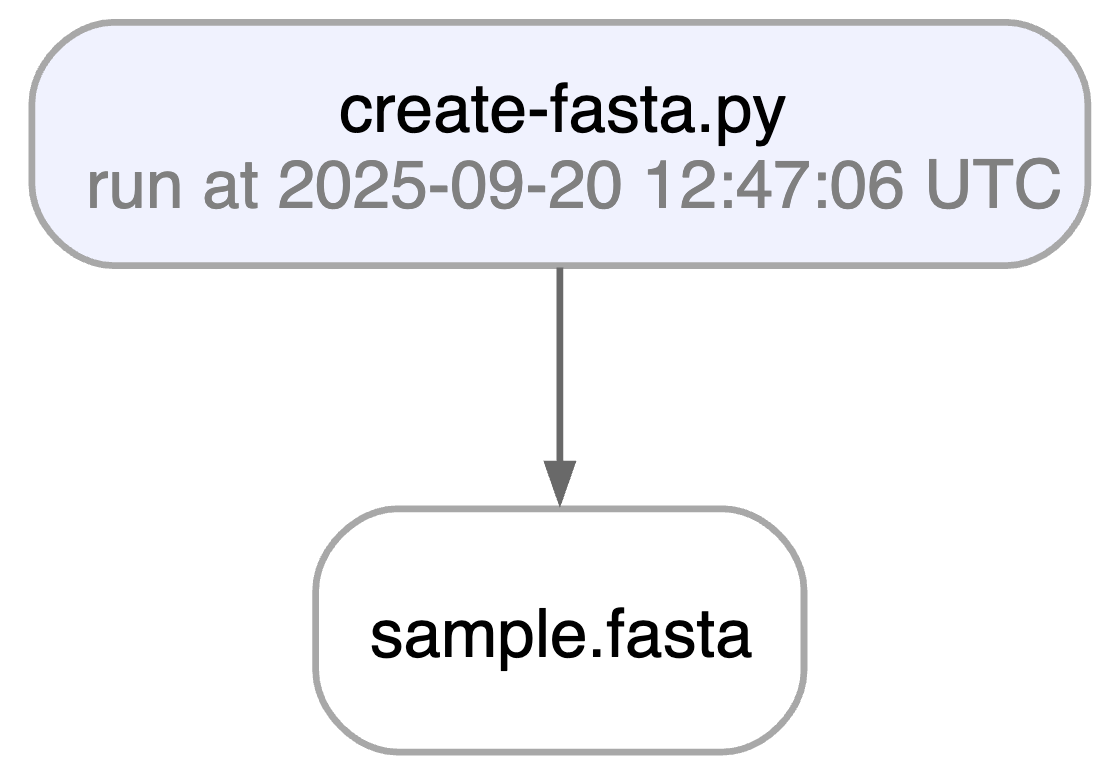
You’ll know how that artifact was created and what it’s used for. Basic metadata was captured in fields:
artifact.size # the file/folder size in bytes
artifact.created_at # the creation timestamp
# etc.
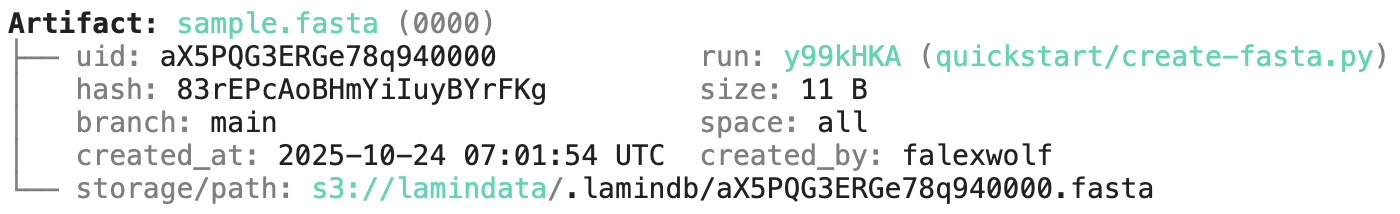
artifact.describe() # describe metadata
Artifact: sample.fasta (0000) ├── uid: Nk3OuRRf4i2gkxp30000 run: lwMrMpW (README.ipynb) │ hash: 83rEPcAoBHmYiIuyBYrFKg size: 11 B │ branch: main space: all │ created_at: 2025-11-27 13:42:59 UTC created_by: anonymous └── storage/path: /home/runner/work/lamindb/lamindb/docs/test-transfer/.lamindb/Nk3OuRRf4i2gkxp30000.fasta
Here is how to access the content of the artifact:
local_path = artifact.cache() # return a local path from a cache
object = artifact.load() # if available for the format, load object into memory
! run input wasn't tracked, call `ln.track()` and re-run
! run input wasn't tracked, call `ln.track()` and re-run
And here is how to access its data lineage context:
run = artifact.run # get the run record
transform = artifact.run.transform # get the transform record
Examples for run & transform.
run.describe()
![]()
Makes your data queryable, traceable, reproducible, and FAIR. One API: lakehouse, lineage, feature store, ontologies, LIMS, ELN.
Why?
Reproducing analytical results or understanding how a dataset or model was created can be a pain. Training models on historical data, LIMS & ELN systems, orthogonal assays, or datasets from other teams is even harder. Even maintaining an overview of a project's datasets & analyses is more difficult than it should be.
Biological datasets are typically managed with versioned storage systems, GUI-focused platforms, structureless data lakes, rigid data warehouses (SQL, monolithic arrays), or tabular lakehouses.
LaminDB extends the lakehouse architecture to biological registries & datasets beyond tables (DataFrame, AnnData, .zarr, .tiledbsoma, …).
It provides enough structure to enable queries across many datasets, enough freedom to keep the pace of R&D high, and rich context in form of data lineage and metadata for humans and AI.
Highlights:
- lineage → track inputs & outputs of notebooks, scripts, functions & pipelines with a single line of code
- lakehouse → manage, monitor & validate schemas; query across many datasets
- feature store → manage features & labels; leverage batch loading
- FAIR datasets → validate & annotate
DataFrame,AnnData,SpatialData,parquet,zarr, … - LIMS & ELN → manage experimental metadata, ontologies & markdown notes
- unified access → storage locations (local, S3, GCP, …), SQL databases (Postgres, SQLite) & ontologies
- reproducible → auto-version & timestamp execution reports, source code & environments
- zero lock-in & scalable → runs in your infrastructure; not a client for a rate-limited REST API
- integrations → vitessce, nextflow, redun, and more
- extendable → create custom plug-ins based on the Django ORM
If you want a GUI: LaminHub is a data collaboration hub built on LaminDB similar to how GitHub is built on git.
Who uses it?
Scientists & engineers in pharma, biotech, and academia, including:
- Pfizer – A global BigPharma company with headquarters in the US
- Ensocell Therapeutics – A BioTech with offices in Cambridge, UK, and California
- DZNE – The National Research Center for Neuro-Degenerative Diseases in Germany
- Helmholtz Munich – The National Research Center for Environmental Health in Germany
- scverse – An international non-profit for open-source omics data tools
- The Global Immunological Swarm Learning Network – Research hospitals at U Bonn, Harvard, MIT, Stanford, ETH Zürich, Charite, Mount Sinai, and others
Docs
Copy summary.md into an LLM chat and let AI explain or read the docs.
Setup
Install the Python package:
pip install lamindb
Create a LaminDB instance:
lamin init --modules bionty --storage ./quickstart-data # or s3://my-bucket, gs://my-bucket
Or if you have write access to an instance, connect to it:
lamin connect account/name
Quickstart
Lineage
Create a dataset while tracking source code, inputs, outputs, logs, and environment:
import lamindb as ln
ln.track() # track execution of source code as a run
open("sample.fasta", "w").write(">seq1\nACGT\n") # create a dataset
ln.Artifact("sample.fasta", key="sample.fasta").save() # save dataset as an artifact
ln.finish() # mark the run as finished
Running this snippet as a script (python create-fasta.py) produces the following data lineage:
artifact = ln.Artifact.get(key="sample.fasta") # get artifact by key
artifact.view_lineage()
You'll know how that artifact was created and what it's used for. Basic metadata was captured in fields:
artifact.size # the file/folder size in bytes
artifact.created_at # the creation timestamp
# etc.
artifact.describe() # describe metadata
Here is how to access the content of the artifact:
local_path = artifact.cache() # return a local path from a cache
object = artifact.load() # if available for the format, load object into memory
And here is how to access its data lineage context:
run = artifact.run # get the run record
transform = artifact.run.transform # get the transform record
Examples for run & transform.
run.describe()
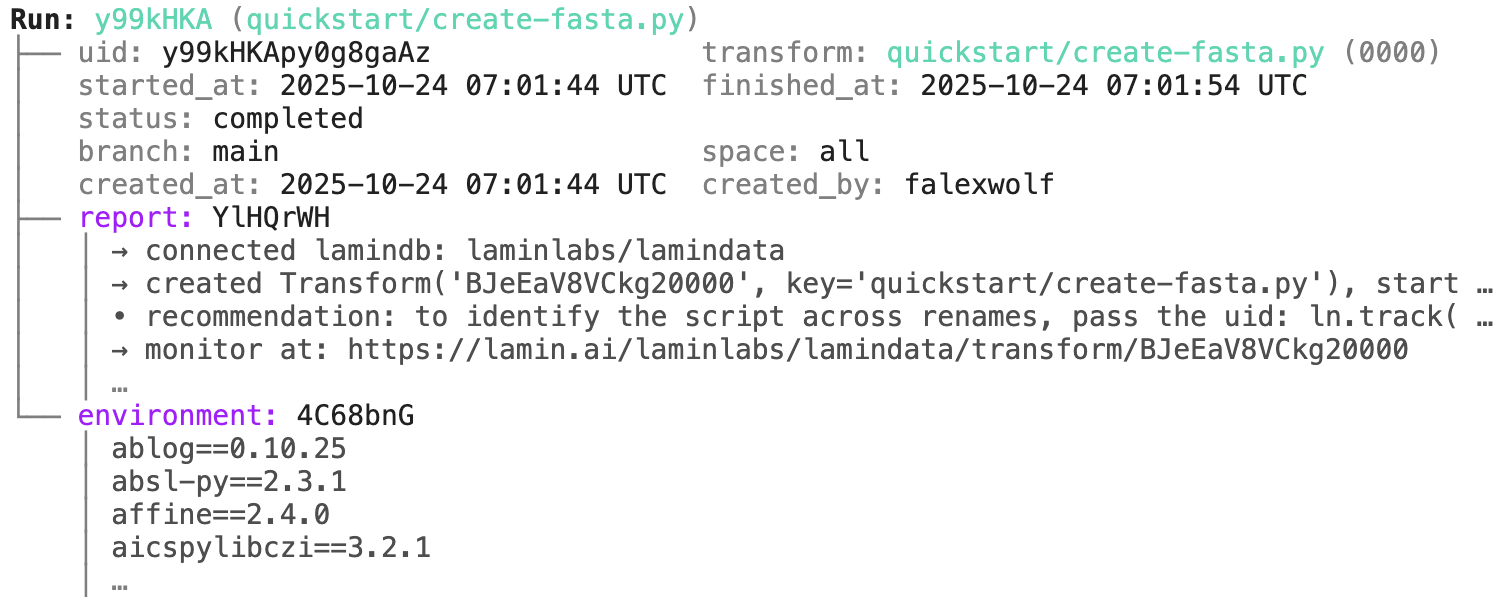
transform.describe()
Sharing
You can share datasets across LaminDB instances.
For example, explore the artifacts in laminlabs/cellxgene. To query & load one that is annotated with Alzheimer's disease:
cellxgene_artifacts = ln.Artifact.connect("laminlabs/cellxgene") # access artifacts in the laminlabs/cellxgene instance
adata = cellxgene_artifacts.get("BnMwC3KZz0BuKftR").load() # load a dataset into memory
from datetime import date
ln.Feature(name="gc_content", dtype=float).save()
ln.Feature(name="experiment_note", dtype=str).save()
ln.Feature(name="experiment_date", dtype=date).save()
During annotation, feature names and data types are validated against these definitions:
artifact.features.add_values({
"gc_content": 0.55,
"experiment_note": "Looks great",
"experiment_date": "2025-10-24",
})
Now that the data is annotated, you can query for it:
ln.Artifact.filter(experiment_date="2025-10-24").to_dataframe() # query all artifacts annotated with `experiment_date`
You can also query by the metadata that lamindb automatically collects:
ln.Artifact.filter(run=run).to_dataframe() # query all artifacts created by a run
ln.Artifact.filter(transform=transform).to_dataframe() # query all artifacts created by a transform
ln.Artifact.filter(size__gt=1e6).to_dataframe() # query all artifacts bigger than 1MB
If you want to include more information into the resulting dataframe, pass include.
ln.Artifact.to_dataframe(include="features") # include the feature annotations
ln.Artifact.to_dataframe(include=["created_by__name", "storage__root"]) # include fields from related registries
sample = ln.Record(name="Sample", is_type=True).save() # type sample
ln.Record(name="P53mutant1", type=sample).save() # sample 1
ln.Record(name="P53mutant2", type=sample).save() # sample 2
Define the corresponding features and annotate:
ln.Feature(name="design_sample", dtype=sample).save()
artifact.features.add_values({"design_sample": "P53mutant1"})
You can query & search the Record registry in the same way as Artifact or Run.
ln.Record.search("p53").to_dataframe()
You can also create relationships of entities and -- if you connect your LaminDB instance to LaminHub -- edit them like Excel sheets in a GUI.
Lake: versioning
If you change source code or datasets, LaminDB manages their versioning for you.
Assume you run a new version of our create-fasta.py script to create a new version of sample.fasta.
import lamindb as ln
ln.track()
open("sample.fasta", "w").write(">seq1\nTGCA\n") # a new sequence
ln.Artifact("sample.fasta", key="sample.fasta", features={"design_sample": "P53mutant1"}).save() # annotate with the new sample
ln.finish()
If you now query by key, you'll get the latest version of this artifact.
artifact = ln.Artifact.get(key="sample.fasta") # get artifact by key
artifact.versions.to_dataframe() # see all versions of that artifact
import pandas as pd
df = pd.DataFrame({
"sequence_str": ["ACGT", "TGCA"],
"gc_content": [0.55, 0.54],
"experiment_note": ["Looks great", "Ok"],
"experiment_date": [date(2025, 10, 24), date(2025, 10, 25)],
})
ln.Artifact.from_dataframe(df, key="my_datasets/sequences.parquet").save() # no validation
To validate & annotate the content of the dataframe, use a built-in schema:
ln.Feature(name="sequence_str", dtype=str).save() # define a remaining feature
artifact = ln.Artifact.from_dataframe(
df,
key="my_datasets/sequences.parquet",
schema="valid_features" # validate columns against features
).save()
artifact.describe()
import anndata as ad
import numpy as np
adata = ad.AnnData(
X=pd.DataFrame([[1]*10]*21).values,
obs=pd.DataFrame({'cell_type_by_model': ['T cell', 'B cell', 'NK cell'] * 7}),
var=pd.DataFrame(index=[f'ENSG{i:011d}' for i in range(10)])
)
artifact = ln.Artifact.from_anndata(
adata,
key="my_datasets/scrna.h5ad",
schema="ensembl_gene_ids_and_valid_features_in_obs"
)
artifact.describe()
To validate a spatialdata or any other array-like dataset, you need to construct a Schema. You can do this by composing the schema of a complicated object from simple pandera/pydantic-like schemas: docs.lamin.ai/curate.
Ontologies
Plugin bionty gives you >20 of them as SQLRecord registries. This was used to validate the ENSG ids in the adata just before.
import bionty as bt
bt.CellType.import_source() # import the default ontology
bt.CellType.to_dataframe() # your extendable cell type ontology in a simple registry
CLI
Most of the functionality that's available in Python is also available on the command line (and in R through LaminR). For instance, to upload a file or folder, run:
lamin save myfile.txt --key examples/myfile.txt
Workflow managers
LaminDB is not a workflow manager, but it integrates well with existing workflow managers and can subsitute them in some settings.
In github.com/laminlabs/schmidt22 we manage several workflows, scripts, and notebooks to re-construct the project of Schmidt el al. (2022). A phenotypic CRISPRa screening result is integrated with scRNA-seq data. Here is one of the input artifacts:
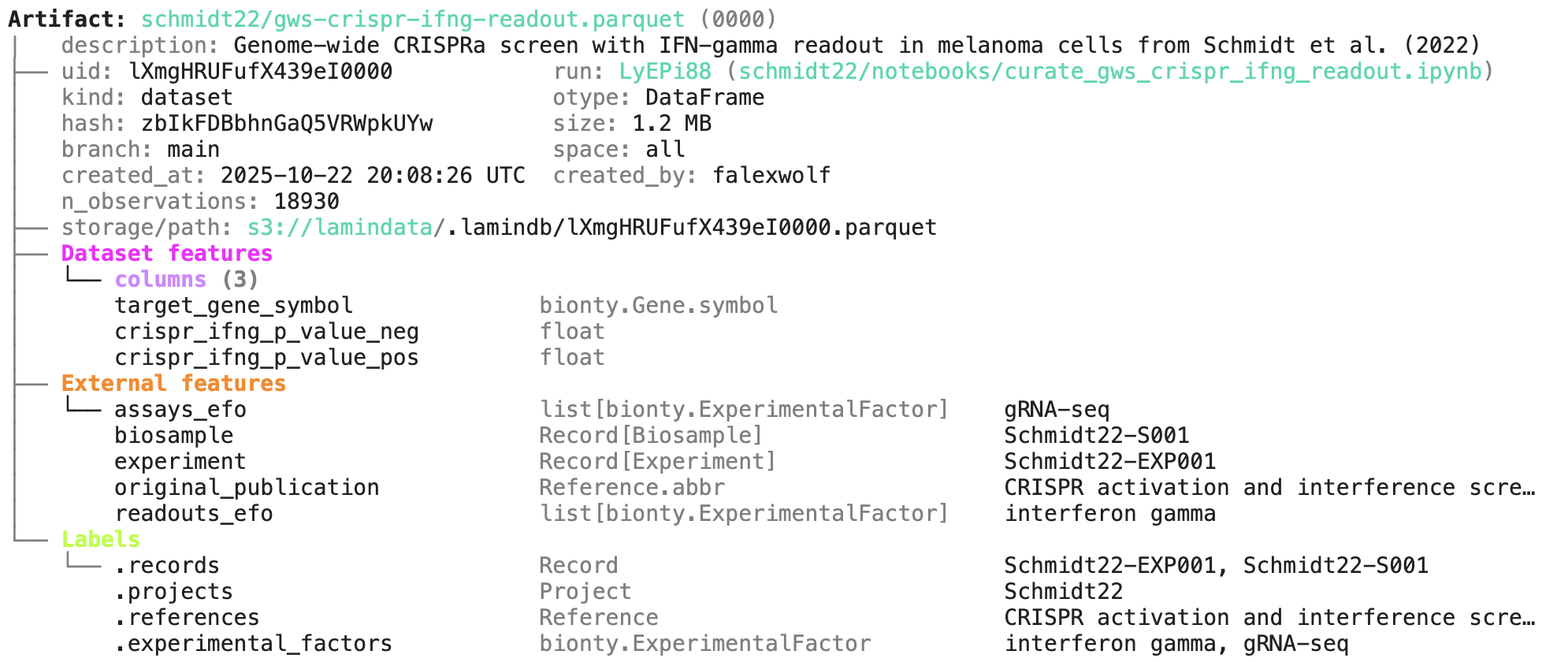
And here is the lineage of the final result:
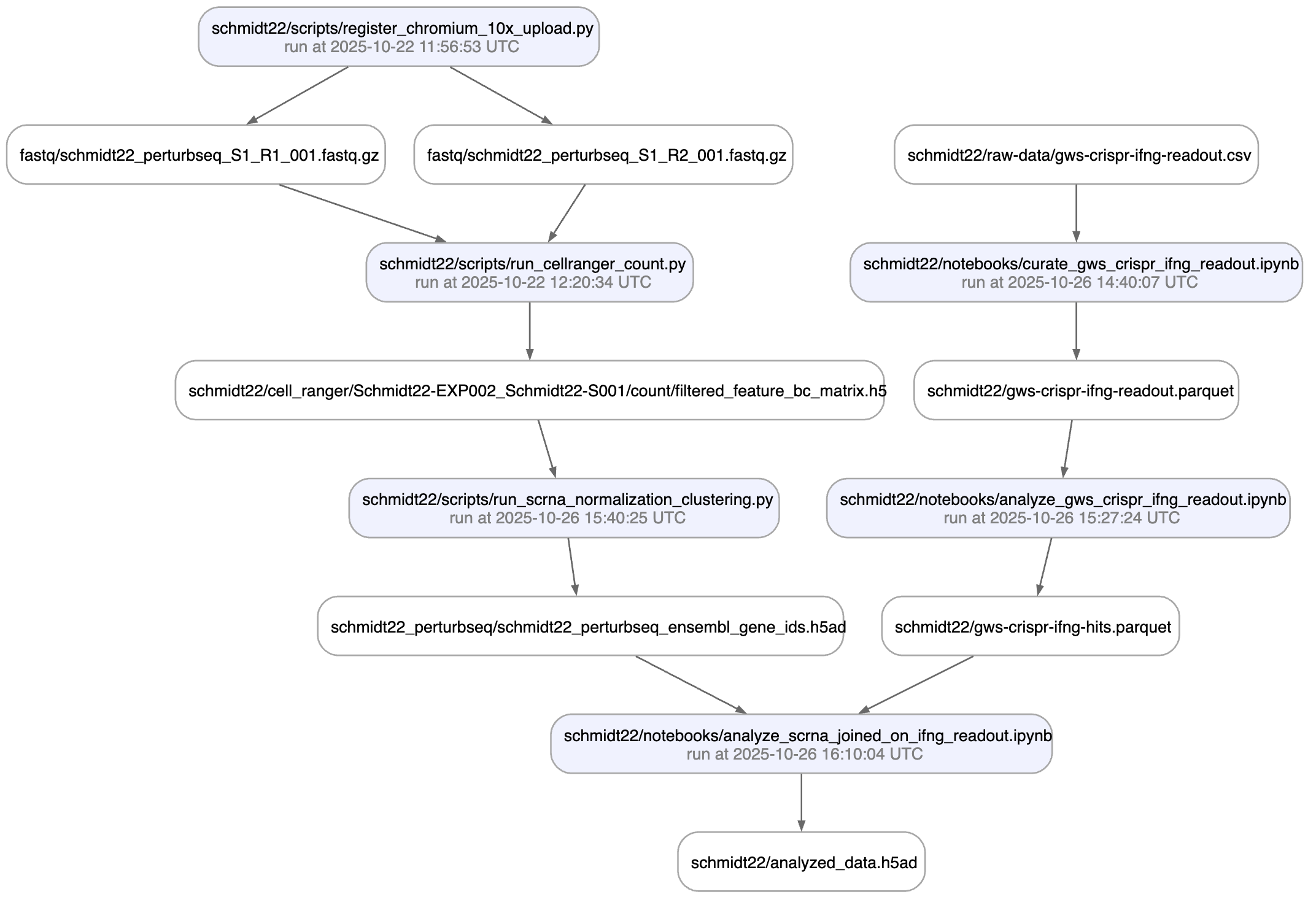
You can explore it here.
If you'd like to integrate with Nextflow, Snakemake, or redun, see here: docs.lamin.ai/pipelines
Run: lwMrMpW (README.ipynb) ├── uid: lwMrMpWqk4EjwXmZ transform: README.ipynb (0000) │ | description: LaminDB - A data framework for biology │ started_at: 2025-11-27 13:42:58 UTC finished_at: 2025-11-27 13:43:01 UTC │ status: completed │ branch: main space: all │ created_at: 2025-11-27 13:42:58 UTC created_by: anonymous └── environment: hSDASzO │ aiobotocore==2.25.2 │ aiohappyeyeballs==2.6.1 │ aiohttp==3.13.2 │ aioitertools==0.13.0 │ …
transform.describe()
Transform: README.ipynb (0000) | description: LaminDB - A data framework for biology ├── uid: 5UreeBqnzjaw0000 │ hash: Coxp803zrhnSqqakyNbBzQ type: notebook │ branch: main space: all │ created_at: 2025-11-27 13:42:58 UTC created_by: anonymous └── source_code: │ # %% [markdown] │ # [](https://docs.lamin. … │ # [](https://docs.lami … │ # [](https:// … │ # │ # │ # │ # Makes your data queryable, traceable, reproducible, and FAIR. One API: lakehou … │ # │ # <details> │ # <summary>Why?</summary> │ # │ # Reproducing analytical results or understanding how a dataset or model was cre … │ # Training models on historical data, LIMS & ELN systems, orthogonal assays, or … │ # Even maintaining an overview of a project's datasets & analyses is more diffic … │ # │ # Biological datasets are typically managed with versioned storage systems, GUI- … │ # │ # LaminDB extends the lakehouse architecture to biological registries & datasets … │ # It provides enough structure to enable queries across many datasets, enough fr … │ # │ # </details> │ # │ # <img width="800px" src="https://lamin-site-assets.s3.amazonaws.com/.lamindb/Bu … │ # │ # Highlights: │ # │ # - **lineage** → track inputs & outputs of notebooks, scripts, functions & pipe … │ …
Lake: annotation & queries¶
You can annotate datasets and samples with features. Let’s define some:
from datetime import date
ln.Feature(name="gc_content", dtype=float).save()
ln.Feature(name="experiment_note", dtype=str).save()
ln.Feature(name="experiment_date", dtype=date).save()
Feature(uid='IfEJ7opFnvnX', name='experiment_date', dtype='date', is_type=None, unit=None, description=None, array_rank=0, array_size=0, array_shape=None, proxy_dtype=None, synonyms=None, branch_id=1, space_id=1, created_by_id=1, run_id=None, type_id=None, created_at=2025-11-27 13:43:09 UTC, is_locked=False)
During annotation, feature names and data types are validated against these definitions:
artifact.features.add_values({
"gc_content": 0.55,
"experiment_note": "Looks great",
"experiment_date": "2025-10-24",
})
Now that the data is annotated, you can query for it:
ln.Artifact.filter(experiment_date="2025-10-24").to_dataframe() # query all artifacts annotated with `experiment_date`
| uid | key | description | suffix | kind | otype | size | hash | n_files | n_observations | params | version | is_latest | is_locked | created_at | branch_id | space_id | storage_id | run_id | schema_id | created_by_id | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| id | |||||||||||||||||||||
| 2 | Nk3OuRRf4i2gkxp30000 | sample.fasta | None | .fasta | None | None | 11 | 83rEPcAoBHmYiIuyBYrFKg | None | None | None | None | True | False | 2025-11-27 13:42:59.628000+00:00 | 1 | 1 | 1 | 3 | None | 1 |
You can also query by the metadata that lamindb automatically collects:
ln.Artifact.filter(run=run).to_dataframe() # query all artifacts created by a run
ln.Artifact.filter(transform=transform).to_dataframe() # query all artifacts created by a transform
ln.Artifact.filter(size__gt=1e6).to_dataframe() # query all artifacts bigger than 1MB
| uid | key | description | suffix | kind | otype | size | hash | n_files | n_observations | params | version | is_latest | is_locked | created_at | branch_id | space_id | storage_id | run_id | schema_id | created_by_id | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| id | |||||||||||||||||||||
| 1 | dPraor9rU1EofcFb6Wph | tabula_sapiens_lung.h5ad | Part of Tabula Sapiens, a benchmark, first-dra... | .h5ad | None | None | 3899435772 | 8mB1KK2wd51F6HQdvqipcQ | None | None | None | None | True | False | 2023-07-14 19:00:30.621330+00:00 | 1 | 1 | 2 | 2 | None | 1 |
If you want to include more information into the resulting dataframe, pass include.
ln.Artifact.to_dataframe(include="features") # include the feature annotations
ln.Artifact.to_dataframe(include=["created_by__name", "storage__root"]) # include fields from related registries
→ queried for all categorical features with dtype Record and non-categorical features: (3) ['gc_content', 'experiment_note', 'experiment_date']
| uid | key | created_by__name | storage__root | |
|---|---|---|---|---|
| id | ||||
| 2 | Nk3OuRRf4i2gkxp30000 | sample.fasta | None | /home/runner/work/lamindb/lamindb/docs/test-tr... |
| 1 | dPraor9rU1EofcFb6Wph | tabula_sapiens_lung.h5ad | None | s3://lamindata |
Lake ♾️ LIMS ♾️ Sheets¶
You can create records for the entities underlying your experiments: samples, perturbations, instruments, etc., for example:
sample = ln.Record(name="Sample", is_type=True).save() # type sample
ln.Record(name="P53mutant1", type=sample).save() # sample 1
ln.Record(name="P53mutant2", type=sample).save() # sample 2
! you are trying to create a record with name='P53mutant2' but a record with similar name exists: 'P53mutant1'. Did you mean to load it?
Record(uid='RtyozA5WTUslOhN0', name='P53mutant2', is_type=False, description=None, reference=None, reference_type=None, params=None, branch_id=1, space_id=1, created_by_id=1, type_id=1, schema_id=None, run_id=None, created_at=2025-11-27 13:43:09 UTC, is_locked=False)
Define the corresponding features and annotate:
ln.Feature(name="design_sample", dtype=sample).save()
artifact.features.add_values({"design_sample": "P53mutant1"})
You can query & search the Record registry in the same way as Artifact or Run.
ln.Record.search("p53").to_dataframe()
| uid | name | is_type | description | reference | reference_type | params | is_locked | created_at | branch_id | space_id | created_by_id | type_id | schema_id | run_id | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| id | |||||||||||||||
| 2 | AYeNDvFyOxx8wfKA | P53mutant1 | False | None | None | None | None | False | 2025-11-27 13:43:09.426000+00:00 | 1 | 1 | 1 | 1 | None | None |
| 3 | RtyozA5WTUslOhN0 | P53mutant2 | False | None | None | None | None | False | 2025-11-27 13:43:09.440000+00:00 | 1 | 1 | 1 | 1 | None | None |
You can also create relationships of entities and – if you connect your LaminDB instance to LaminHub – edit them like Excel sheets in a GUI.
Lake: versioning¶
If you change source code or datasets, LaminDB manages their versioning for you.
Assume you run a new version of our create-fasta.py script to create a new version of sample.fasta.
import lamindb as ln
ln.track()
open("sample.fasta", "w").write(">seq1\nTGCA\n") # a new sequence
ln.Artifact("sample.fasta", key="sample.fasta", features={"design_sample": "P53mutant1"}).save() # annotate with the new sample
ln.finish()
→ found notebook README.ipynb, making new version -- anticipating changes
→ created Transform('5UreeBqnzjaw0001', key='README.ipynb'), started new Run('fl2O53LGwdDcNVGH') at 2025-11-27 13:43:09 UTC
→ notebook imports: anndata==0.12.2 bionty==1.9.1 lamindb==1.17a1 numpy==2.3.5 pandas==2.3.3
• recommendation: to identify the notebook across renames, pass the uid: ln.track("5UreeBqnzjaw")
→ creating new artifact version for key 'sample.fasta' in storage '/home/runner/work/lamindb/lamindb/docs/test-transfer'
! cells [(0, 2), (16, 18)] were not run consecutively
→ returning artifact with same hash: Artifact(uid='h2YDyMrDOc0b0vX00000', version=None, is_latest=True, key=None, description='Report of run lwMrMpWqk4EjwXmZ', suffix='.html', kind='__lamindb_run__', otype=None, size=319315, hash='h2qjr6-QlIPPr5wK0SI-PQ', n_files=None, n_observations=None, params=None, branch_id=1, space_id=1, storage_id=1, run_id=None, schema_id=None, created_by_id=1, created_at=2025-11-27 13:43:01 UTC, is_locked=False); to track this artifact as an input, use: ln.Artifact.get()
! run was not set on Artifact(uid='h2YDyMrDOc0b0vX00000', version=None, is_latest=True, key=None, description='Report of run lwMrMpWqk4EjwXmZ', suffix='.html', kind='__lamindb_run__', otype=None, size=319315, hash='h2qjr6-QlIPPr5wK0SI-PQ', n_files=None, n_observations=None, params=None, branch_id=1, space_id=1, storage_id=1, run_id=None, schema_id=None, created_by_id=1, created_at=2025-11-27 13:43:01 UTC, is_locked=False), setting to current run
! updated description from Report of run lwMrMpWqk4EjwXmZ to Report of run fl2O53LGwdDcNVGH
! returning transform with same hash & key: Transform(uid='5UreeBqnzjaw0000', version=None, is_latest=False, key='README.ipynb', description='LaminDB - A data framework for biology', type='notebook', hash='Coxp803zrhnSqqakyNbBzQ', reference=None, reference_type=None, config=None, is_flow=False, flow=None, environment=None, branch_id=1, space_id=1, created_by_id=1, created_at=2025-11-27 13:42:58 UTC, is_locked=False)
• new latest version is: Transform(uid='5UreeBqnzjaw0000', version=None, is_latest=True, key='README.ipynb', description='LaminDB - A data framework for biology', type='notebook', hash='Coxp803zrhnSqqakyNbBzQ', reference=None, reference_type=None, config=None, is_flow=False, flow=None, environment=None, branch_id=1, space_id=1, created_by_id=1, created_at=2025-11-27 13:42:58 UTC, is_locked=False)
→ finished Run('fl2O53LGwdDcNVGH') after 1s at 2025-11-27 13:43:11 UTC
If you now query by key, you’ll get the latest version of this artifact.
artifact = ln.Artifact.get(key="sample.fasta") # get artifact by key
artifact.versions.to_dataframe() # see all versions of that artifact
| uid | key | description | suffix | kind | otype | size | hash | n_files | n_observations | params | version | is_latest | is_locked | created_at | branch_id | space_id | storage_id | run_id | schema_id | created_by_id | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| id | |||||||||||||||||||||
| 5 | Nk3OuRRf4i2gkxp30001 | sample.fasta | None | .fasta | None | None | 11 | aqvq4CskQu3Nnr3hl5r3ug | None | None | None | None | True | False | 2025-11-27 13:43:10.429000+00:00 | 1 | 1 | 1 | 4 | None | 1 |
| 2 | Nk3OuRRf4i2gkxp30000 | sample.fasta | None | .fasta | None | None | 11 | 83rEPcAoBHmYiIuyBYrFKg | None | None | None | None | False | False | 2025-11-27 13:42:59.628000+00:00 | 1 | 1 | 1 | 3 | None | 1 |
Lakehouse ♾️ feature store¶
Here is how you ingest a DataFrame:
import pandas as pd
df = pd.DataFrame({
"sequence_str": ["ACGT", "TGCA"],
"gc_content": [0.55, 0.54],
"experiment_note": ["Looks great", "Ok"],
"experiment_date": [date(2025, 10, 24), date(2025, 10, 25)],
})
ln.Artifact.from_dataframe(df, key="my_datasets/sequences.parquet").save() # no validation
→ writing the in-memory object into cache
Artifact(uid='jlJwJuRI8zbH1rPU0000', version=None, is_latest=True, key='my_datasets/sequences.parquet', description=None, suffix='.parquet', kind='dataset', otype='DataFrame', size=3405, hash='K3fBSY6wjjJr9VlKBhymJw', n_files=None, n_observations=2, params=None, branch_id=1, space_id=1, storage_id=1, run_id=None, schema_id=None, created_by_id=1, created_at=2025-11-27 13:43:11 UTC, is_locked=False)
To validate & annotate the content of the dataframe, use a built-in schema:
ln.Feature(name="sequence_str", dtype=str).save() # define a remaining feature
artifact = ln.Artifact.from_dataframe(
df,
key="my_datasets/sequences.parquet",
schema="valid_features" # validate columns against features
).save()
artifact.describe()
→ writing the in-memory object into cache
→ returning artifact with same hash: Artifact(uid='jlJwJuRI8zbH1rPU0000', version=None, is_latest=True, key='my_datasets/sequences.parquet', description=None, suffix='.parquet', kind='dataset', otype='DataFrame', size=3405, hash='K3fBSY6wjjJr9VlKBhymJw', n_files=None, n_observations=2, params=None, branch_id=1, space_id=1, storage_id=1, run_id=None, schema_id=None, created_by_id=1, created_at=2025-11-27 13:43:11 UTC, is_locked=False); to track this artifact as an input, use: ln.Artifact.get()
→ loading artifact into memory for validation
✓ "columns" is validated against Feature.name
Artifact: my_datasets/sequences.parquet (0000) ├── uid: jlJwJuRI8zbH1rPU0000 run: │ kind: dataset otype: DataFrame │ hash: K3fBSY6wjjJr9VlKBhymJw size: 3.3 KB │ branch: main space: all │ created_at: 2025-11-27 13:43:11 UTC created_by: anonymous │ n_observations: 2 ├── storage/path: /home/runner/work/lamindb/lamindb/docs/test-transfer/.lamindb/jlJwJuRI8zbH1rPU0000.parquet └── Dataset features └── columns (4) gc_content float experiment_note str experiment_date date sequence_str str
Now you know which schema the dataset satisfies. You can filter for datasets by schema and then launch distributed queries and batch loading.
Lakehouse beyond tables¶
To validate an AnnData with a built-in schema call:
import anndata as ad
import numpy as np
adata = ad.AnnData(
X=pd.DataFrame([[1]*10]*21).values,
obs=pd.DataFrame({'cell_type_by_model': ['T cell', 'B cell', 'NK cell'] * 7}),
var=pd.DataFrame(index=[f'ENSG{i:011d}' for i in range(10)])
)
artifact = ln.Artifact.from_anndata(
adata,
key="my_datasets/scrna.h5ad",
schema="ensembl_gene_ids_and_valid_features_in_obs"
)
artifact.describe()
→ writing the in-memory object into cache
→ loading artifact into memory for validation
! 1 term not validated in feature 'columns' in slot 'obs': 'cell_type_by_model'
→ fix typos, remove non-existent values, or save terms via: curator.slots['obs'].cat.add_new_from('columns')
! no values were validated for columns!
/opt/hostedtoolcache/Python/3.13.9/x64/lib/python3.13/functools.py:934: ImplicitModificationWarning: Transforming to str index.
return dispatch(args[0].__class__)(*args, **kw)
✓ created 1 Organism record from Bionty matching name: 'human'
✓ added 2 records from_public with bionty.Gene for "columns": 'ENSG00000000003', 'ENSG00000000005'
! 8 terms not validated in feature 'columns' in slot 'var.T': 'ENSG00000000000', 'ENSG00000000001', 'ENSG00000000002', 'ENSG00000000004', 'ENSG00000000006', 'ENSG00000000007', 'ENSG00000000008', 'ENSG00000000009'
→ fix organism 'human', fix typos, remove non-existent values, or save terms via: curator.slots['var.T'].cat.add_new_from('columns')
Artifact: my_datasets/scrna.h5ad (0000) ├── uid: 7ZRq02wQozZO1UYs0000 run: │ kind: dataset otype: AnnData │ hash: Sgbj2aSf8AKFs12oJKUxXQ size: 20.9 KB │ branch: main space: all │ created_at: <django.db.models.expressions.DatabaseDefault object at 0x7fe0845956d0> created_by: anonymous │ n_observations: 21 └── storage/path: /home/runner/work/lamindb/lamindb/docs/test-transfer/.lamindb/7ZRq02wQozZO1UYs0000.h5ad
To validate a spatialdata or any other array-like dataset, you need to construct a Schema. You can do this by composing the schema of a complicated object from simple pandera/pydantic-like schemas: docs.lamin.ai/curate.
Ontologies¶
Plugin bionty gives you >20 of them as SQLRecord registries. This was used to validate the ENSG ids in the adata just before.
import bionty as bt
bt.CellType.import_source() # import the default ontology
bt.CellType.to_dataframe() # your extendable cell type ontology in a simple registry
✓ import is completed!
| uid | name | ontology_id | abbr | synonyms | description | is_locked | created_at | branch_id | space_id | created_by_id | run_id | source_id | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| id | |||||||||||||
| 3209 | 6nw5na3P | lateral pyloric motor neuron | CL:4070017 | None | LP | A Motor Neuron That Controls Pyloric Filter Mo... | False | 2025-11-27 13:43:19.201000+00:00 | 1 | 1 | 1 | None | 16 |
| 3208 | 7k6v66wY | anterior burster neuron | CL:4070016 | None | AB | Pyloric Pacemaker Neuron That Provides Feedbac... | False | 2025-11-27 13:43:19.201000+00:00 | 1 | 1 | 1 | None | 16 |
| 3207 | 2YBgl1kN | pyloric dilator neuron | CL:4070015 | None | PD | A Motor Neuron That Controls The Cardiopyloric... | False | 2025-11-27 13:43:19.201000+00:00 | 1 | 1 | 1 | None | 16 |
| 3206 | 4FhOIxSq | pyloric motor neuron | CL:4070014 | None | PY | A Motor Neuron That Controls Pyloric Filter Mo... | False | 2025-11-27 13:43:19.201000+00:00 | 1 | 1 | 1 | None | 16 |
| 3205 | 7N982L0j | ventricular dilator motor neuron | CL:4070013 | None | VD | A Motor Neuron That Controls Ventral Stomach ... | False | 2025-11-27 13:43:19.201000+00:00 | 1 | 1 | 1 | None | 16 |
| ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... |
| 3114 | 1lYpfaRM | middle primary motoneuron | CL:4042041 | None | None | A Type Of Primary Motor Neuron Located In The ... | False | 2025-11-27 13:43:19.188000+00:00 | 1 | 1 | 1 | None | 16 |
| 3113 | 4wsKKFDu | glutamatergic neuron of the basal ganglia | CL:4042040 | None | None | A Glutametergic Neuron With Its Soma Located I... | False | 2025-11-27 13:43:19.188000+00:00 | 1 | 1 | 1 | None | 16 |
| 3112 | 2GneiPp3 | caudal ganglionic eminence derived neuron | CL:4042039 | None | None | A Neuron Of The Central Nervous System That De... | False | 2025-11-27 13:43:19.188000+00:00 | 1 | 1 | 1 | None | 16 |
| 3111 | 3bfpC22i | rostral primary motorneuron | CL:4042038 | None | None | A Type Of Primary Motor Neuron Situated In The... | False | 2025-11-27 13:43:19.188000+00:00 | 1 | 1 | 1 | None | 16 |
| 3110 | 3zrwm9Cz | sst GABAergic neuron of the striatum | CL:4042037 | None | None | A Interneuron Of The Striatum Expressing Gamma... | False | 2025-11-27 13:43:19.188000+00:00 | 1 | 1 | 1 | None | 16 |
100 rows × 13 columns
CLI¶
Most of the functionality that’s available in Python is also available on the command line (and in R through LaminR). For instance, to upload a file or folder, run:
lamin save myfile.txt --key examples/myfile.txt
Workflow managers¶
LaminDB is not a workflow manager, but it integrates well with existing workflow managers and can subsitute them in some settings.
In github.com/laminlabs/schmidt22 we manage several workflows, scripts, and notebooks to re-construct the project of Schmidt el al. (2022). A phenotypic CRISPRa screening result is integrated with scRNA-seq data. Here is one of the input artifacts:
And here is the lineage of the final result:
You can explore it here.
If you’d like to integrate with Nextflow, Snakemake, or redun, see here: docs.lamin.ai/pipelines